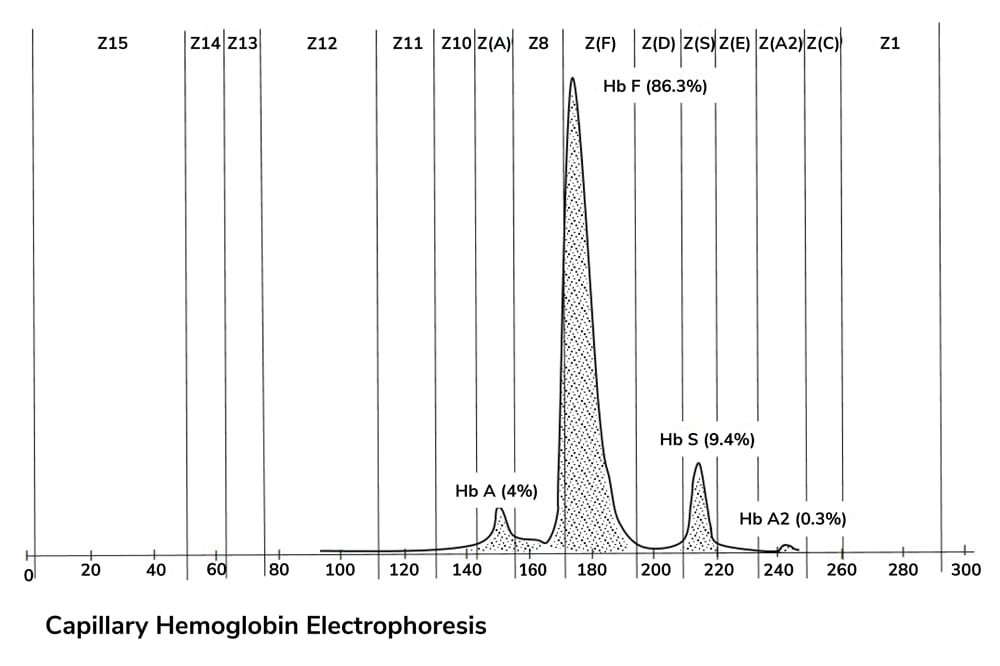
A study published in Immunity provides new insight into how chronic intestinal inflammation in inflammatory bowel disease (IBD) may contribute to the development of colorectal cancer. The research focuses on immune signaling pathways that link colitis to increased neutrophil production and tumor-associated inflammation.
Patients with long-standing IBD are known to have a higher risk of colorectal cancer, but the biological mechanisms underlying this association are not fully defined. In this study, researchers examined the role of tumor necrosis factor–like cytokine 1A (TL1A), an inflammatory cytokine previously linked to severe IBD, in shaping immune responses in the colon.
Using mouse models of colitis and colitis-associated cancer, along with analyses of human intestinal tissue, the investigators found that TL1A activates a subset of innate immune cells called type 3 innate lymphoid cells (ILC3s). When stimulated by TL1A, these cells produced granulocyte–macrophage colony-stimulating factor (GM-CSF), which in turn promoted the rapid production of neutrophils in the bone marrow, a process known as emergency granulopoiesis.
The neutrophils generated during chronic inflammation differed from those typically involved in acute infection. Gene expression analysis showed that they shared features with tumor-associated neutrophils, including markers linked to inflammation, tissue remodeling, and tumor growth. Spatial analysis of colon tissue revealed that these neutrophils accumulated near dysplastic and cancerous lesions rather than in normal areas of the colon.
In mouse models, blocking TL1A signaling or reducing neutrophil numbers led to a decrease in tumor burden, suggesting that this inflammatory pathway contributes directly to cancer development in the context of colitis. While these findings were generated primarily in experimental systems, the authors also analyzed human samples to assess clinical relevance.
In biopsies from patients with IBD, dysplastic lesions showed increased expression of neutrophil-related gene signatures associated with tumor-promoting activity compared with sporadic colorectal dysplasia. In addition, samples from patients with ulcerative colitis treated with anti-TL1A therapy showed reduced expression of several neutrophil-associated genes after treatment.
Overall, this study helps clarify how persistent immune activation in IBD may shift normal inflammatory responses toward a tumor-promoting environment. It also highlights diagnostic challenges, including distinguishing harmful neutrophil populations from protective ones and interpreting immune gene signatures in chronically inflamed tissue.
Although the study does not establish new diagnostic markers, it provides a mechanistic framework that may inform future efforts to assess cancer risk in patients with IBD using immune and spatial profiling approaches.
Newsletters
Receive the latest pathologist news, personalities, education, and career development – weekly to your inbox.